multiomics
[ ]:
[1]:
#!pip install muon scanpy hvplot statsmodels scikit-learn
[7]:
pip install -e ../../STREAM2/
Obtaining file:///mnt/c/Users/jobac/Desktop/all/git/STREAM2
Preparing metadata (setup.py) ... done
Requirement already satisfied: numpy>=1.17.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (1.23.5)
Requirement already satisfied: numba>=0.52.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.52.0)
Requirement already satisfied: networkx>=2.5 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (3.1)
Requirement already satisfied: pandas!=1.1,>=1.0 in /home/jo/.local/lib/python3.8/site-packages (from stream2==0.1a0) (1.3.5)
Requirement already satisfied: anndata>=0.7.4 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.8.0)
Requirement already satisfied: scanpy>=1.6.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (1.9.3)
Requirement already satisfied: shapely>=2.0.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (2.0.1)
Requirement already satisfied: statsmodels>=0.12.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.13.5)
Requirement already satisfied: scikit-learn>=1.2 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (1.2.2)
Requirement already satisfied: scipy>=1.4 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (1.10.1)
Requirement already satisfied: kneed>=0.7 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.7.0)
Requirement already satisfied: seaborn>=0.11 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.12.2)
Requirement already satisfied: matplotlib>=3.3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (3.7.1)
Requirement already satisfied: plotly>=4.14.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (5.14.1)
Requirement already satisfied: scikit-misc>=0.1.3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.2.0)
Requirement already satisfied: adjusttext>=0.7.3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.8)
Requirement already satisfied: umap-learn>=0.4.6 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.5.3)
Requirement already satisfied: elpigraph-python>=0.3.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (0.3.1)
Requirement already satisfied: python-slugify>=5.0.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from stream2==0.1a0) (8.0.1)
Requirement already satisfied: h5py>=3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from anndata>=0.7.4->stream2==0.1a0) (3.8.0)
Requirement already satisfied: natsort in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from anndata>=0.7.4->stream2==0.1a0) (8.3.1)
Requirement already satisfied: packaging>=20 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from anndata>=0.7.4->stream2==0.1a0) (23.1)
Requirement already satisfied: python-igraph>=0.7.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from elpigraph-python>=0.3.1->stream2==0.1a0) (0.10.4)
Requirement already satisfied: contourpy>=1.0.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (1.0.7)
Requirement already satisfied: cycler>=0.10 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (0.11.0)
Requirement already satisfied: fonttools>=4.22.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (4.39.3)
Requirement already satisfied: kiwisolver>=1.0.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (1.4.4)
Requirement already satisfied: pillow>=6.2.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (9.4.0)
Requirement already satisfied: pyparsing>=2.3.1 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (3.0.9)
Requirement already satisfied: python-dateutil>=2.7 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (2.8.2)
Requirement already satisfied: importlib-resources>=3.2.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from matplotlib>=3.3->stream2==0.1a0) (5.12.0)
Requirement already satisfied: llvmlite<0.36,>=0.35.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from numba>=0.52.0->stream2==0.1a0) (0.35.0)
Requirement already satisfied: setuptools in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from numba>=0.52.0->stream2==0.1a0) (67.6.1)
Requirement already satisfied: pytz>=2017.3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from pandas!=1.1,>=1.0->stream2==0.1a0) (2023.3)
Requirement already satisfied: tenacity>=6.2.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from plotly>=4.14.0->stream2==0.1a0) (8.2.2)
Requirement already satisfied: text-unidecode>=1.3 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from python-slugify>=5.0.0->stream2==0.1a0) (1.3)
Requirement already satisfied: tqdm in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from scanpy>=1.6.0->stream2==0.1a0) (4.65.0)
Requirement already satisfied: patsy in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from scanpy>=1.6.0->stream2==0.1a0) (0.5.3)
Requirement already satisfied: joblib in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from scanpy>=1.6.0->stream2==0.1a0) (1.2.0)
Requirement already satisfied: session-info in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from scanpy>=1.6.0->stream2==0.1a0) (1.0.0)
Requirement already satisfied: threadpoolctl>=2.0.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from scikit-learn>=1.2->stream2==0.1a0) (3.1.0)
Requirement already satisfied: pynndescent>=0.5 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from umap-learn>=0.4.6->stream2==0.1a0) (0.5.10)
Requirement already satisfied: zipp>=3.1.0 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from importlib-resources>=3.2.0->matplotlib>=3.3->stream2==0.1a0) (3.15.0)
Requirement already satisfied: six in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from patsy->scanpy>=1.6.0->stream2==0.1a0) (1.16.0)
Requirement already satisfied: igraph==0.10.4 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from python-igraph>=0.7.1->elpigraph-python>=0.3.1->stream2==0.1a0) (0.10.4)
Requirement already satisfied: texttable>=1.6.2 in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from igraph==0.10.4->python-igraph>=0.7.1->elpigraph-python>=0.3.1->stream2==0.1a0) (1.6.7)
Requirement already satisfied: stdlib-list in /home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages (from session-info->scanpy>=1.6.0->stream2==0.1a0) (0.8.0)
Installing collected packages: stream2
Attempting uninstall: stream2
Found existing installation: stream2 0.1a0
Uninstalling stream2-0.1a0:
Successfully uninstalled stream2-0.1a0
Running setup.py develop for stream2
Successfully installed stream2-0.1a0
Note: you may need to restart the kernel to use updated packages.
[1]:
import scanpy as sc
import muon as mu
import stream2 as st2
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
import hvplot.pandas
from session_info import show
show()
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
---------------------------------------------------------------------------
KeyError Traceback (most recent call last)
Cell In[1], line 11
9 import hvplot.pandas
10 from session_info import show
---> 11 show()
File ~/anaconda3/envs/stream2/lib/python3.8/site-packages/session_info/main.py:209, in show(na, os, cpu, jupyter, dependencies, std_lib, private, write_req_file, req_file_name, html, excludes)
207 for mod_name in clean_modules:
208 mod_names.append(mod_name)
--> 209 mod = sys.modules[mod_name]
210 # Since modules use different attribute names to store version info,
211 # try the most common ones.
212 try:
KeyError: 'cupy'
Multi-modal trajectory inference demo
We load four assays (two scATAC and two scRNA) integrated into one anndata object.
adata_integrated.X contains gene expression (gene scores for ATAC cells) for 200 highly variable genes.
We will use this subset of measurements from hematopoietic cells as a toy example.
[2]:
adata_integrated = st2.read_h5ad('../data/multiomics/atac_rna_toy200.h5ad')
#sc.pp.neighbors(adata_integrated,n_neighbors=50,metric='cosine',use_rep='X_pca_transp_emd')
#sc.tl.umap(adata_integrated,min_dist=0.05)
sc.pl.umap(adata_integrated[::-1],color=['assay_detailed','leiden_identity_simple2',],wspace=.2)
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/anndata/compat/_overloaded_dict.py:106: ImplicitModificationWarning: Trying to modify attribute `._uns` of view, initializing view as actual.
self.data[key] = value
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(

In this case we have 4 assays with same features (since we only loaded gene scores for ATAC), which fits nicely into a single anndata.
However in general it is not possible to simply concatenate assays with possibly different features and observations into a single anndata.
The solution is to use the muon package and its MuData format to store our multimodal assays
[3]:
assays = adata_integrated.obs['assay_detailed']
adatas = {assay: adata_integrated[assays==assay] for assay in assays.unique()}
mdata = mu.MuData(adatas)
mdata
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/mudata/_core/mudata.py:457: UserWarning: Cannot join columns with the same name because var_names are intersecting.
warnings.warn(
[3]:
MuData object with n_obs × n_vars = 99955 × 800
4 modalities
ATAC_CD: 13575 x 200
obs: 'leiden_identity_simple2', 'label', 'assay_detailed'
obsm: 'X_pca_transp_emd', 'X_umap'
ATAC_days: 19206 x 200
obs: 'leiden_identity_simple2', 'label', 'assay_detailed'
obsm: 'X_pca_transp_emd', 'X_umap'
RNA_CD: 22804 x 200
obs: 'leiden_identity_simple2', 'label', 'assay_detailed'
obsm: 'X_pca_transp_emd', 'X_umap'
RNA_days: 44370 x 200
obs: 'leiden_identity_simple2', 'label', 'assay_detailed'
obsm: 'X_pca_transp_emd', 'X_umap'In our case cells are not matched between assays so we had to perform data integration to get a joint UMAP representation We will learn a joint trajectory based on this joint representation. Let’s store it along with cell annotation at the global rather than assay level in mdata
[4]:
mdata.obsm['X_dr'] = mdata.obsm['X_umap'] = np.vstack([adata.obsm['X_umap'] for adata in mdata.mod.values()])
mdata.obsm['X_pca_transp_emd'] = np.vstack([adata.obsm['X_pca_transp_emd'] for adata in mdata.mod.values()])
mdata.obs['leiden_identity_simple2'] = np.hstack([adata.obs['leiden_identity_simple2'] for adata in mdata.mod.values()])
If your data contains matched cells, you have more choice and can learn the trajectory based on: - some RNA representation - some ATAC representation - some joint RNA+ATAC representation obtained using your favorite method e.g.: - MOFA (mu.tl.mofa(mdata)) - J-UMAP https://github.com/canzarlab/JVis-learn - UMAP tutorial for multiple views https://umap-learn.readthedocs.io/en/latest/composing_models.html
If your data contains non-matched cells then assays can be integrated into a joint representation. Some suggestions of methods to obtain it: - Harmony, Scanorama (scanpy.pp.external) - Optimal transport https://pythonot.github.io/ - Joint MDS https://github.com/BorgwardtLab/JointMDS - MultiMAP https://github.com/Teichlab/MultiMAP
Let’s work with a subsample (just to speed up trajectory inference, it’s also fine to use mdata directly)
[5]:
idx = np.random.RandomState(0).choice(mdata.shape[0],5000,replace=False)
a_s = sc.AnnData(np.zeros((5000,1)))
a_s.obsm['X_pca_transp_emd'] = mdata.obsm['X_pca_transp_emd'][idx]
a_s.obsm['X_dr'] = a_s.obsm['X_umap'] = mdata.obsm['X_umap'][idx]
a_s.obs['leiden_identity_simple2'] = mdata.obs['leiden_identity_simple2'][idx].values
/tmp/ipykernel_2000/1930611853.py:3: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
a_s = sc.AnnData(np.zeros((5000,1)))
Trajectory inference in 2d
We will first illustrate how to learn a trajectory in 2D before illustrating higher dimensions.
We seed graph with a minimum spanning tree. Forbid the tree to form paths connecting Neu-like to DC-like monocytes
[12]:
st2.tl.seed_graph(a_s,n_clusters=80, label_strength=2.5,
paths_disfavored=[['Neu-like monocytes','DC-like monocytes']],label='leiden_identity_simple2')
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5),key='seed_epg')
Seeding initial graph...
Clustering...
K-Means clustering ...
Calculating minimum spanning tree...
/mnt/c/Users/jobac/Desktop/all/git/STREAM2/stream2/tools/_elpigraph.py:865: FutureWarning: Unlike other reduction functions (e.g. `skew`, `kurtosis`), the default behavior of `mode` typically preserves the axis it acts along. In SciPy 1.11.0, this behavior will change: the default value of `keepdims` will become False, the `axis` over which the statistic is taken will be eliminated, and the value None will no longer be accepted. Set `keepdims` to True or False to avoid this warning.
modes[i] = scipy.stats.mode(labels[part == i]).mode[0]
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/scipy/stats/_stats_py.py:112: RuntimeWarning: The input array could not be properly checked for nan values. nan values will be ignored.
warnings.warn("The input array could not be properly "
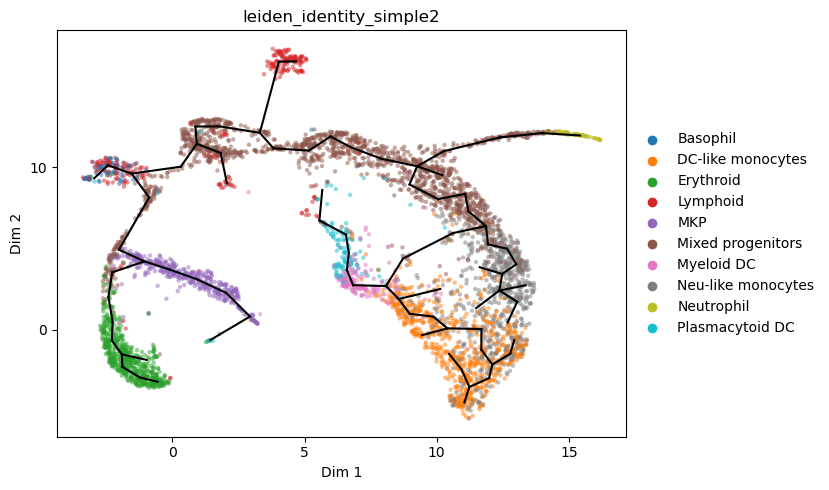
We refine the graph.
We speed up computations by limiting the max number of candidate graphs considered when fitting.
We emphasize less dense regions with slightly higher point weights
[13]:
st2.tl.get_weights(a_s,bandwidth=2)
st2.tl.learn_graph(a_s,n_nodes=95,epg_lambda=0.02,epg_mu=0.05,epg_alpha=0.02,use_weights=True,verbose=1,
max_candidates = {"AddNode2Node": 10,"BisectEdge": 10,"ShrinkEdge": 0,})
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(10,6))
Constructing tree 1 of 1 / Subset 1 of 1
Computing EPG with 95 nodes on 5000 points and 2 dimensions
BARCODE ENERGY NNODES NEDGES NRIBS NSTARS NRAYS NRAYS2 MSE MSEP FVE FVEP UE UR URN URN2 URSD
1|13||81 1.8126 81 80 50 13 0 0 0.504 0.454 0.9915 0.9924 1.2937 0.0148 1.1968 96.9448 0
1|12||82 1.7496 82 81 53 12 0 0 0.5022 0.4546 0.9916 0.9924 1.2332 0.0143 1.1693 95.8848 0
1|11||83 1.6986 83 82 56 11 0 0 0.5013 0.4559 0.9916 0.9923 1.1829 0.0144 1.1959 99.2564 0
12||84 1.6596 84 83 58 12 0 0 0.492 0.4479 0.9917 0.9925 1.1546 0.0131 1.0982 92.2525 0
11||85 1.6202 85 84 61 11 0 0 0.4905 0.4492 0.9918 0.9925 1.1164 0.0133 1.1298 96.0343 0
10||86 1.5849 86 85 64 10 0 0 0.4853 0.4457 0.9918 0.9925 1.0868 0.0128 1.1006 94.6513 0
9||87 1.5572 87 86 67 9 0 0 0.4834 0.4453 0.9919 0.9925 1.0609 0.0129 1.1238 97.7669 0
9||88 1.5259 88 87 68 9 0 0 0.4748 0.438 0.992 0.9926 1.0391 0.0119 1.046 92.046 0
8||89 1.4977 89 88 71 8 0 0 0.4675 0.432 0.9921 0.9927 1.019 0.0112 0.9961 88.656 0
8||90 1.4824 90 89 72 8 0 0 0.4763 0.4411 0.992 0.9926 0.9918 0.0143 1.2884 115.9605 0
7||91 1.4582 91 90 75 7 0 0 0.4765 0.4426 0.992 0.9926 0.9693 0.0124 1.1254 102.415 0
7||92 1.4527 92 91 76 7 0 0 0.4855 0.453 0.9918 0.9924 0.9506 0.0166 1.5287 140.639 0
7||93 1.4382 93 92 77 7 0 0 0.4822 0.4507 0.9919 0.9924 0.9394 0.0166 1.5483 143.9908 0
7||94 1.4305 94 93 78 7 0 0 0.4874 0.456 0.9918 0.9923 0.9256 0.0175 1.6493 155.031 0
7||95 1.417 95 94 79 7 0 0 0.4883 0.4581 0.9918 0.9923 0.9113 0.0173 1.6421 156.0022 0
36.4012 seconds elapsed
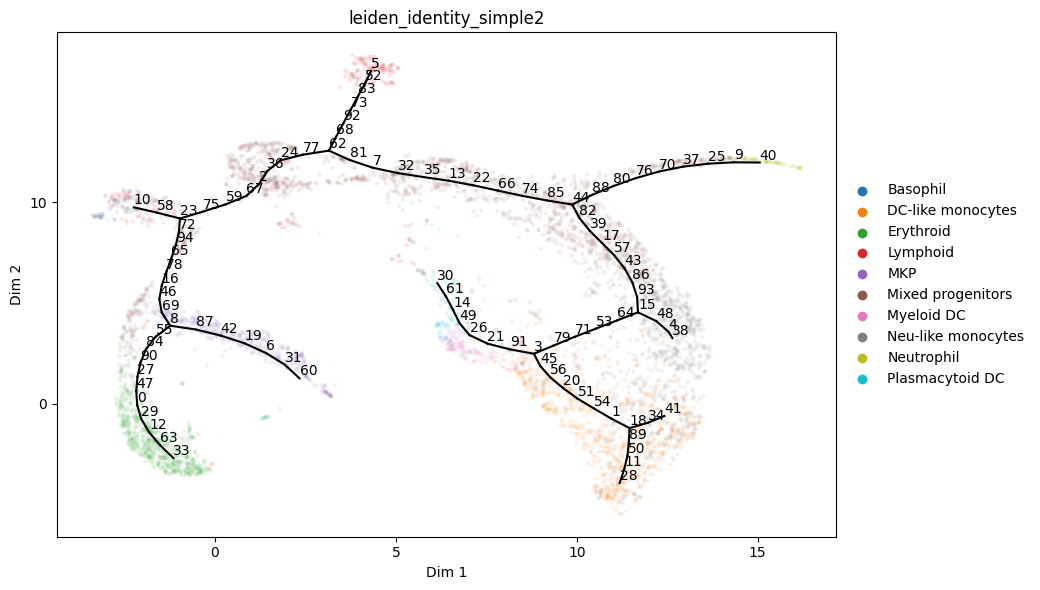
Visualize node labels
[10]:
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.1,size=10,fig_size=(10,6),show_text=1)
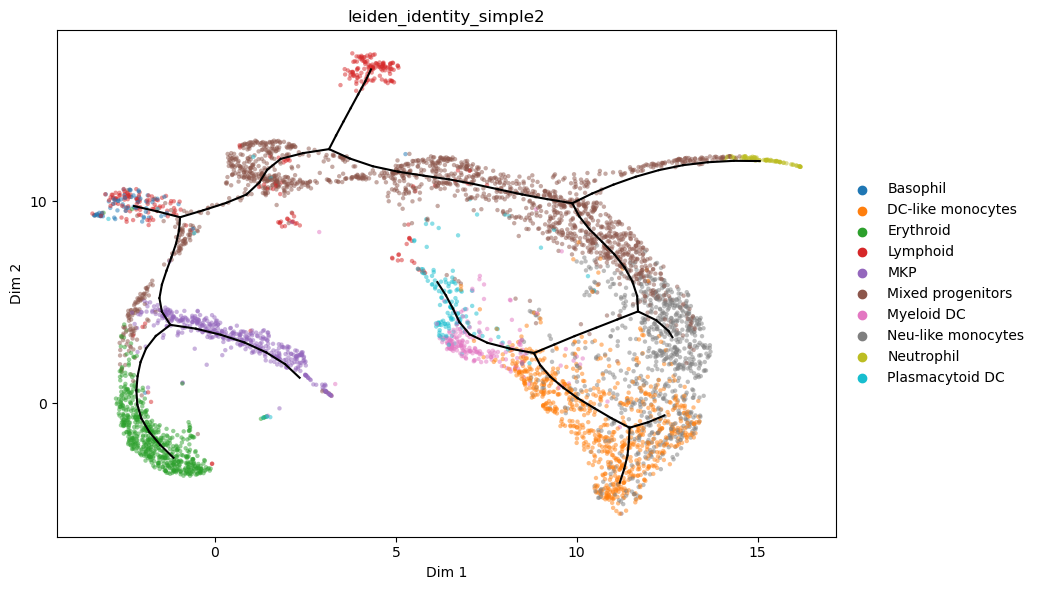
Fine tune the graph by: - adding extra plausible paths - refitting the graph while forcing some nodes to remain fixed at chosen positions - extending leaves so branches reach the end of the distribution
[11]:
a_s2 = a_s.copy()
st2.tl.add_path(a_s2,38,41,refit_graph=True,use_weights=True)
st2.tl.refit_graph(a_s2,shift_nodes_pos={
3 : a_s2.uns['epg']['node_pos'][3] + np.array([+2, +0]),
24 : a_s2.uns['epg']['node_pos'][24] + np.array([-.6, +.2]),
25 : a_s2.uns['epg']['node_pos'][25] + np.array([+0., +.2]),
8 : a_s2.uns['epg']['node_pos'][8] + np.array([-1, +.75]),
44 : a_s2.uns['epg']['node_pos'][44] + np.array([-.8,+.8]),
}
)
st2.tl.add_path(a_s2,2,32,epg_lambda=.2)
st2.tl.add_path(a_s2,30,35,epg_lambda=.2,epg_mu=.2)
st2.tl.add_path(a_s2,99,[2.,9.],epg_lambda=.2,epg_mu=.2)
st2.tl.extend_leaves(a_s2,Mode="QuantDists",ControlPar=.75)
st2.pl.graph(a_s2,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5))
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
(7, 2)
(112, 2)
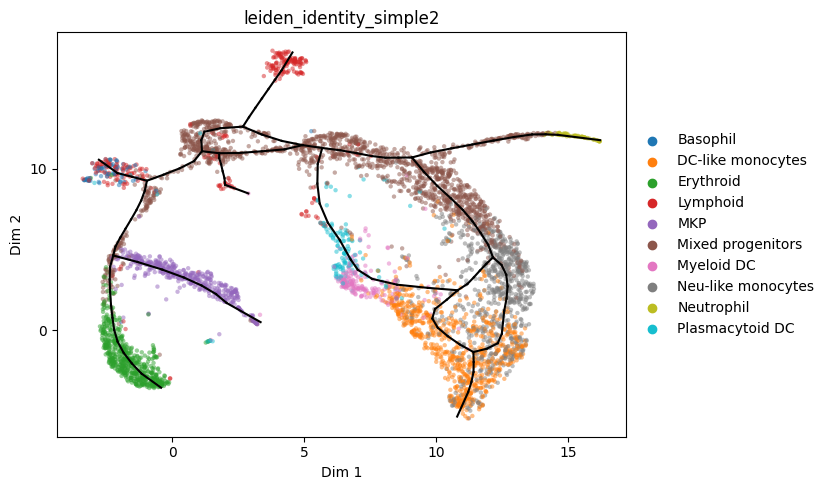
Store graph computed on the subsample back to mdata with the full dataset
[16]:
mdata.uns = a_s2.uns.copy()
# this recomputes the projection of cells onto the graph
st2.tl._elpigraph._store_graph_attributes(mdata,mdata.obsm['X_dr'],'epg')
Infer pseudotime for a given root node
[17]:
st2.tl.infer_pseudotime(mdata,source=2)
st2.pl.graph(mdata,color=['epg_pseudotime'],size=.1,alpha=.3)
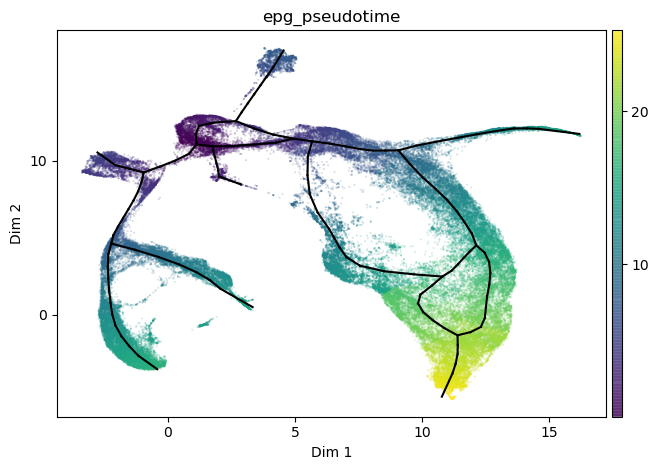
Infer pseudotime for a given path
[18]:
st2.tl.infer_pseudotime(mdata,source=66,target=1)
st2.pl.graph(mdata,color=['epg_pseudotime'],size=.1,alpha=.3)
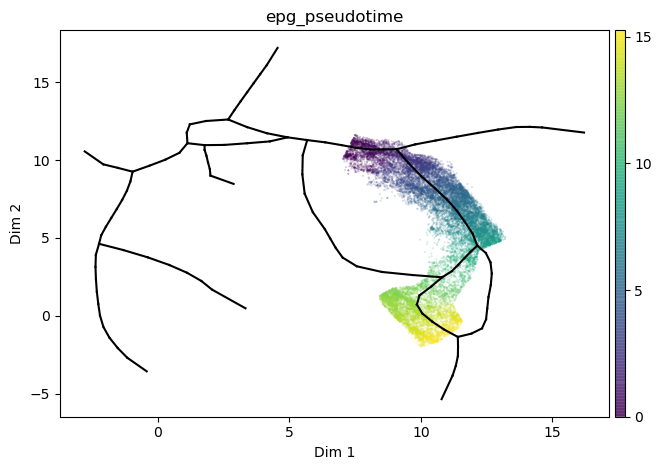
Use nodes_to_include to take a longer path rather than the shortest (default)
[19]:
st2.tl.infer_pseudotime(mdata,source=66,target=1,nodes_to_include=[26])
st2.pl.graph(mdata,color=['epg_pseudotime'],size=.1,alpha=.3)
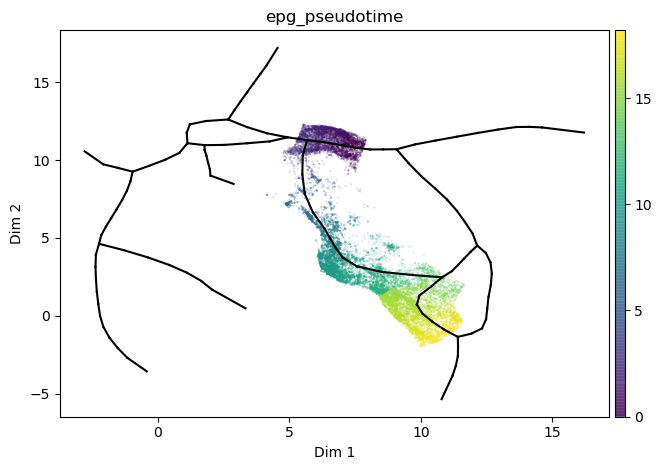
Let’s investigate Erythroids. We compute transition markers (that go up or down along the path) for each assay separately
method = ‘spearman’ for spearman correlation
method = ‘xi’ for non-linear XI correlation, returns values between 0-1
[20]:
source, target = 24, 115
st2.tl.infer_pseudotime(mdata,source=24,target=115)
st2.pl.graph(mdata,color=['epg_pseudotime'],size=.1,alpha=.3)
transition_markers = {}
for assay,adata in mdata.mod.items():
adata.uns['epg']=mdata.uns['epg']
adata.obs = adata.obs.join(mdata.obs[[k for k in mdata.obs if 'epg' in k]])
st2.tl.infer_pseudotime(adata,source=24,target=115)
st2.tl.detect_transition_markers(adata,fc_cutoff=0., method='spearman')
transition_markers[assay] = adata.uns['transition_markers'][f'Path_{source}-None-{target}']
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/anndata/compat/_overloaded_dict.py:106: ImplicitModificationWarning: Trying to modify attribute `._uns` of view, initializing view as actual.
self.data[key] = value
2905 Cells are selected for Path_Source_Nodes-to-include_Target : Path_24-None-115
Filtering out markers that are expressed in less than 5 cells ...
200 markers are being scanned ...
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/anndata/compat/_overloaded_dict.py:106: ImplicitModificationWarning: Trying to modify attribute `._uns` of view, initializing view as actual.
self.data[key] = value
3508 Cells are selected for Path_Source_Nodes-to-include_Target : Path_24-None-115
Filtering out markers that are expressed in less than 5 cells ...
200 markers are being scanned ...
3993 Cells are selected for Path_Source_Nodes-to-include_Target : Path_24-None-115
Filtering out markers that are expressed in less than 5 cells ...
200 markers are being scanned ...
9514 Cells are selected for Path_Source_Nodes-to-include_Target : Path_24-None-115
Filtering out markers that are expressed in less than 5 cells ...
200 markers are being scanned ...
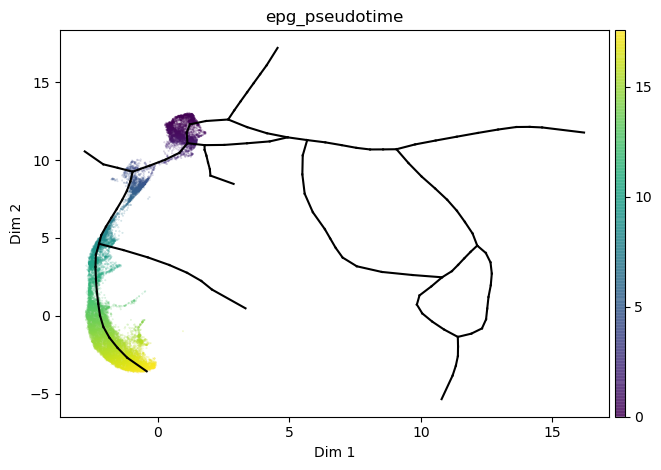
Plot results
[21]:
def plot_top_markers(transition_markers,sort_by = 'logfc',n_genes = 25):
f,axs=plt.subplots(2,2,figsize=(10,10),sharex=True)
axs=axs.flat
for i,n in zip(range(len(transition_markers)),transition_markers.keys()):
# get data
df = transition_markers[n][sort_by].sort_values()[::-1][:n_genes]
gene_names, scores = df.index, df.values
# plot
ymax, ymin = np.max(scores), np.min(scores) - 0.05*np.min(scores)
ymax += 0.3 * (ymax - ymin)
ax=next(axs)
ax.set_ylim(ymin, ymax)
ax.set_xlim(-0.9, n_genes - 0.1)
ax.set_title(n,fontsize=25)
if i>1: ax.set_xlabel('ranking',fontsize=20)
if i==0 or i==2: ax.set_ylabel(sort_by,fontsize=20)
# Making labels
for ig, gene_name in enumerate(gene_names):
ax.text(ig,scores[ig],gene_name,rotation='vertical',verticalalignment='bottom',horizontalalignment='center',fontsize=10,)
plt.tight_layout(); plt.show()
plot_top_markers(transition_markers, sort_by = 'stat',n_genes = 25,)
df = pd.concat(transition_markers).reset_index().rename(columns={'level_0':'assay_detailed','level_1':'var_names'})
df.hvplot.scatter("stat", "logfc", flip_yaxis=False, logy=False, by='assay_detailed',hover_cols=["var_names"],height=500,width=800)
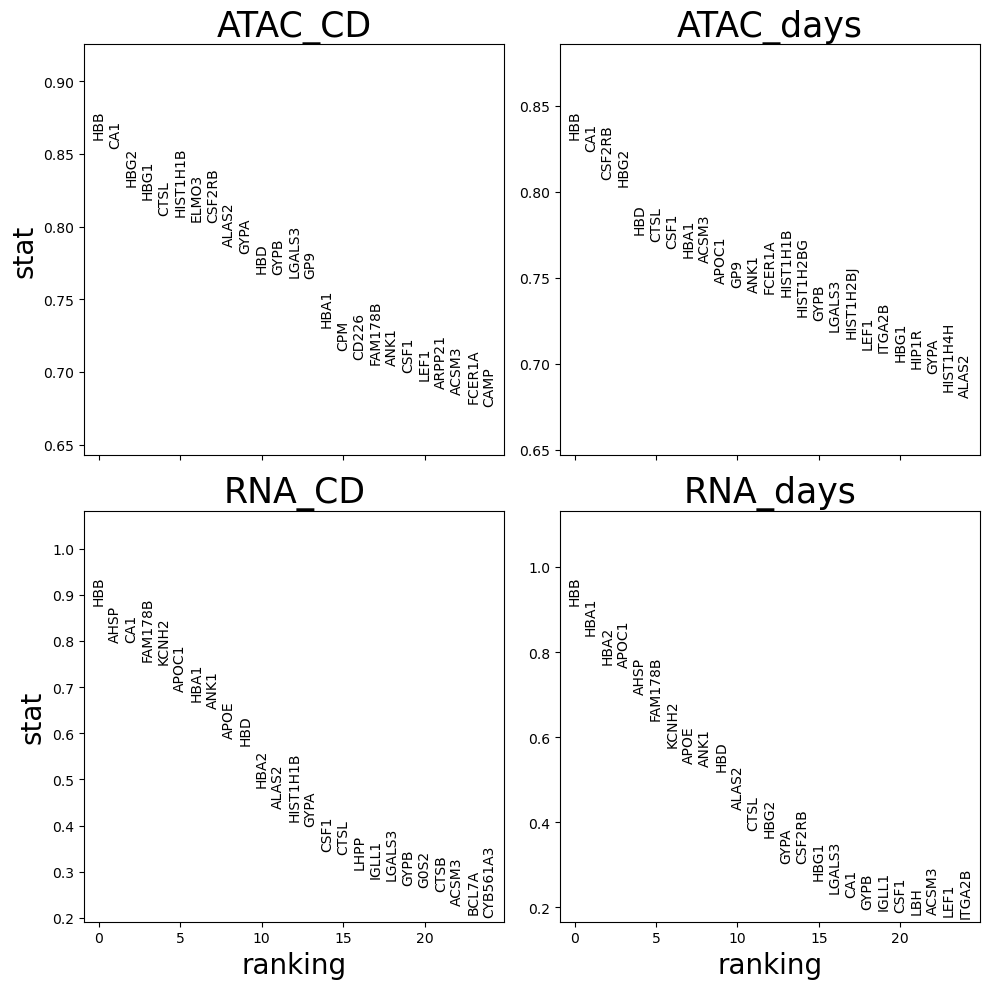
[21]:
Let’s check some of those markers found in several assays
[22]:
def scatterplot_assays(color):
f,axs=plt.subplots(2,2,figsize=(10,10),sharex=True)
axs=axs.flat
for assay,adata in mdata.mod.items():
ax=next(axs)
sc.pl.umap(adata,color=color,ax=ax,show=False)
ax.set_title(assay)
plt.show()
scatterplot_assays('HBB')
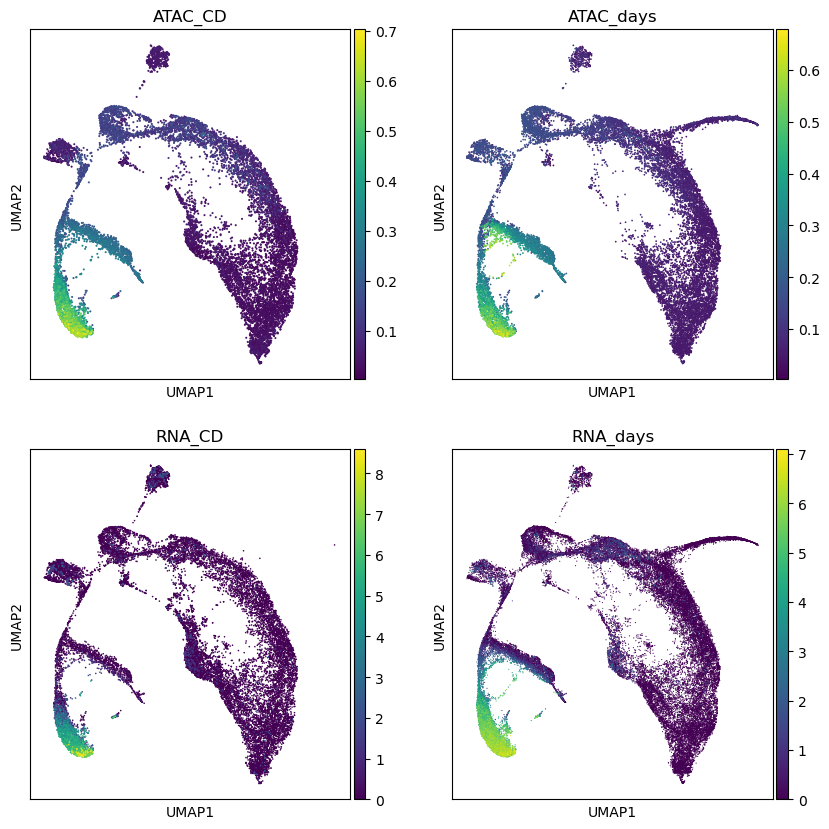
We can try to correlate assays’ variables directly.
In our case cells are not matched across assays; this means they will have different pseudotime values
To deal with this a simple approach is to interpolate data and resample at the same pseudotime values. If your cells are matched you don’t need this step
[23]:
import scipy
interpolated = {}
for assay, adata in mdata.mod.items():
#arrays need to be dense for this step
if scipy.sparse.issparse(adata.X):
adata.X=adata.X.toarray()
interpolated[assay] = st2.tl.interpolate(adata,t_len=200,method='knn',frac=.1,n_neighbors='auto',weights='uniform')[1]
Now that we have data with the same pseudotime values we can correlate variables with same name across assays
[24]:
correlations = {}
modalities = list(mdata.mod.keys())
for i in range(mdata.n_mod-1):
for j in range(i+1,mdata.n_mod):
correlations[modalities[i],modalities[j]] = st2.tl.spearman_columns(interpolated[modalities[i]],
interpolated[modalities[j]])
#or st2.tl.xicorr_columns()[0]
correlations = pd.DataFrame(correlations)
correlations.index = mdata[modalities[0]].var_names
correlations.columns = correlations.columns.to_flat_index()
sns.clustermap(correlations.dropna(),vmin=-1,vmax=1)
[24]:
<seaborn.matrix.ClusterGrid at 0x7f6142416970>
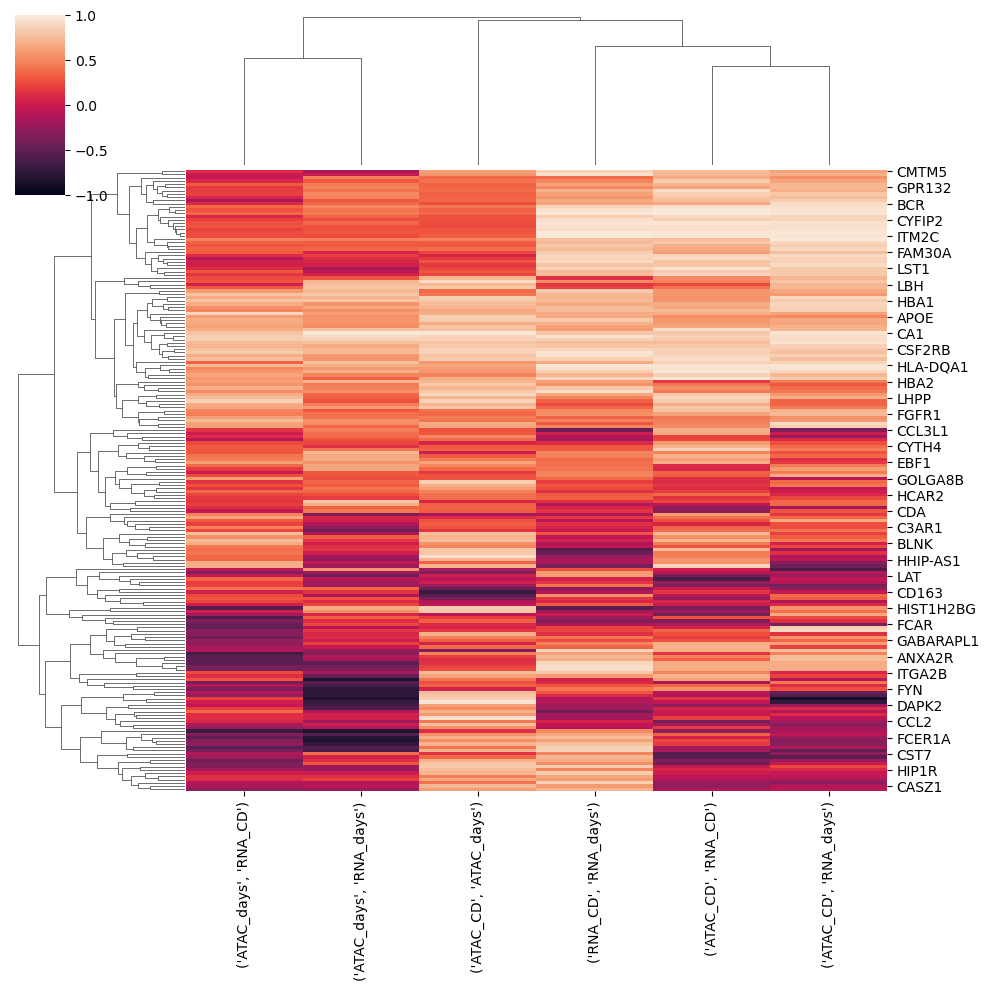
If variables were different across assays we could correlate all with all instead
[25]:
correlations_all = {}
modalities = list(mdata.mod.keys())
for i in range(mdata.n_mod-1):
for j in range(i+1,mdata.n_mod):
correlations_all[modalities[i],modalities[j]] = st2.tl.spearman_pairwise(interpolated[modalities[i]],
interpolated[modalities[j]])
#or st2.tl.xicorr_pairwise()[0]
mi,mj = 'RNA_CD','RNA_days'
corrs_all = pd.DataFrame(correlations_all[mi,mj])
corrs_all.index, corrs_all.columns = mdata[mi].var_names, mdata[mj].var_names
corrs_all[corrs_all.isna()]=0.
sns.clustermap(corrs_all,vmin=-1,vmax=1)
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/seaborn/matrix.py:560: UserWarning: Clustering large matrix with scipy. Installing `fastcluster` may give better performance.
warnings.warn(msg)
[25]:
<seaborn.matrix.ClusterGrid at 0x7f6142c8e160>
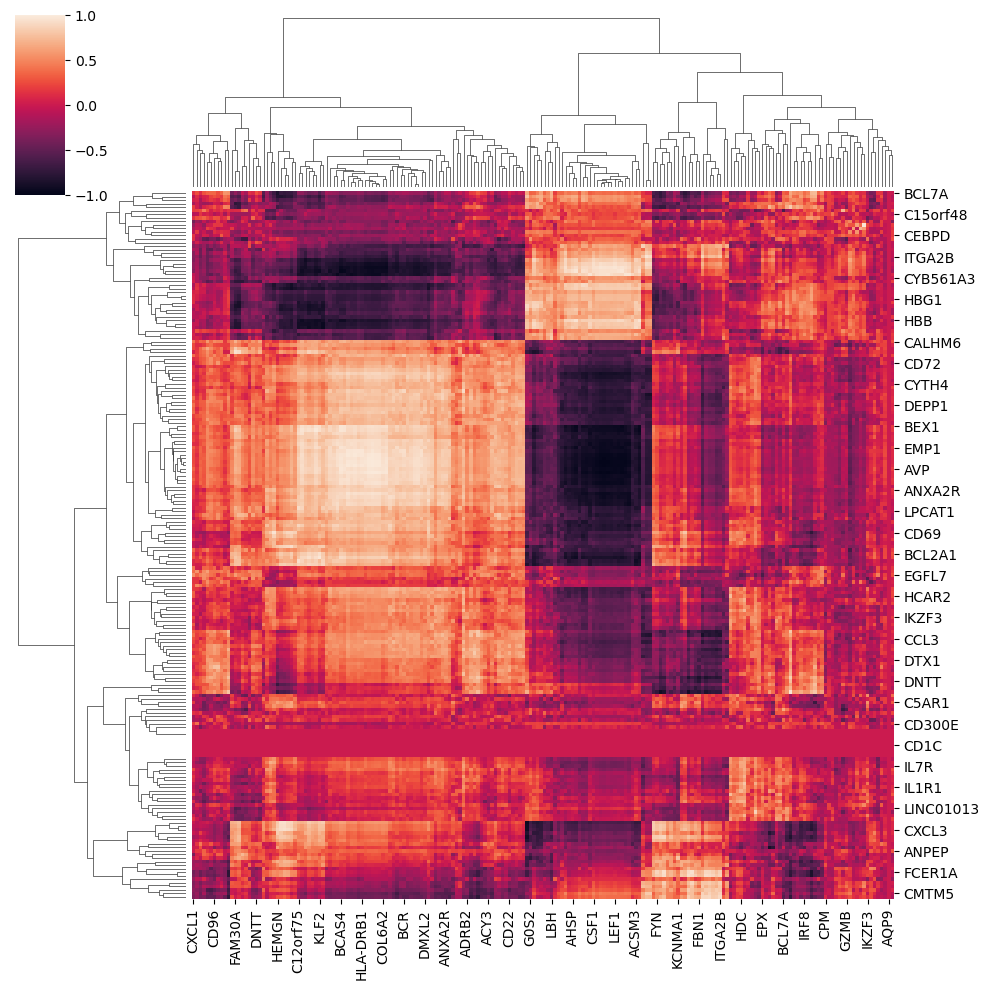
Let’s visualize results as scatter plots.
e.g., we can plot spearman correlation against pseudotime for two assays and color by their correlation.
[26]:
def plot_two_assays(transition_markers,mod1, mod2, plot_var='stat',color='dodgerblue',cmap='Viridis'):
selected = [mod1,mod2]
selected_dfs = {k:v for k,v in transition_markers.items() if k in selected}
df = pd.concat(selected_dfs,axis=1,keys=selected).dropna()
df.columns = df.columns.to_flat_index().map(lambda s:' '.join(s))
if type(color)!=str:
df = df.join(color)
color, clabel = df[color.name], color.name
else:
clabel = color
df.index.name='var_name'
return df.hvplot.scatter(f'{mod1} {plot_var}',f'{mod2} {plot_var}', hover_cols=["var_name"],
color=color,cmap=cmap,clabel=clabel, hover=True,
xlim=(-1.05,1.05),ylim=(-1.05,1.05),height=500,width=600)
def compose_fig(plots,ncols=2):
figure = '('+ '+'.join([f'plots[{i}]' for i in range(len(plots))]) + f').cols({ncols})'
return eval(figure)
plots = []
for i in range(mdata.n_mod-1):
for j in range(i+1,mdata.n_mod):
color = correlations[modalities[i],modalities[j]].rename('correlation '+modalities[i]+' vs '+modalities[j])
plots.append( plot_two_assays(transition_markers,modalities[i],modalities[j], plot_var='stat', color=color) )
compose_fig(plots,2)
[26]:
Trajectory inference in higher dimension
We now show how the trajectory can be learnt based on the higher dimensional PCA data instead
[27]:
a_s.obsm['X_dr'] = a_s.obsm['X_pca_transp_emd']
st2.tl.seed_graph(a_s,n_clusters=80,label_strength=2.5,paths_disfavored=[['Neu-like monocytes','DC-like monocytes']],label='leiden_identity_simple2')
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5),key='seed_epg')
Seeding initial graph...
Clustering...
K-Means clustering ...
Calculating minimum spanning tree...
/mnt/c/Users/jobac/Desktop/all/git/STREAM2/stream2/tools/_elpigraph.py:865: FutureWarning: Unlike other reduction functions (e.g. `skew`, `kurtosis`), the default behavior of `mode` typically preserves the axis it acts along. In SciPy 1.11.0, this behavior will change: the default value of `keepdims` will become False, the `axis` over which the statistic is taken will be eliminated, and the value None will no longer be accepted. Set `keepdims` to True or False to avoid this warning.
modes[i] = scipy.stats.mode(labels[part == i]).mode[0]
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/scipy/stats/_stats_py.py:112: RuntimeWarning: The input array could not be properly checked for nan values. nan values will be ignored.
warnings.warn("The input array could not be properly "
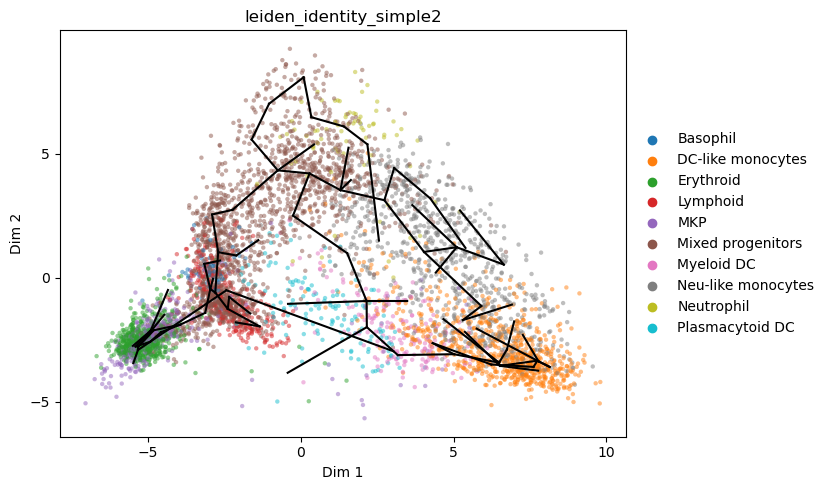
To make sense of how the graph fits to data, it can be useful to roughly represent its position on the 2D umap rather than PC axes.
We achieve this by projecting data points onto their closest node, and defining 2D node positions as the centroid of their associated points
[28]:
st2.tl.project_graph(a_s, to_basis='X_umap', key='seed_epg')
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5),key='seed_epg_from_X_dr_to_X_umap')
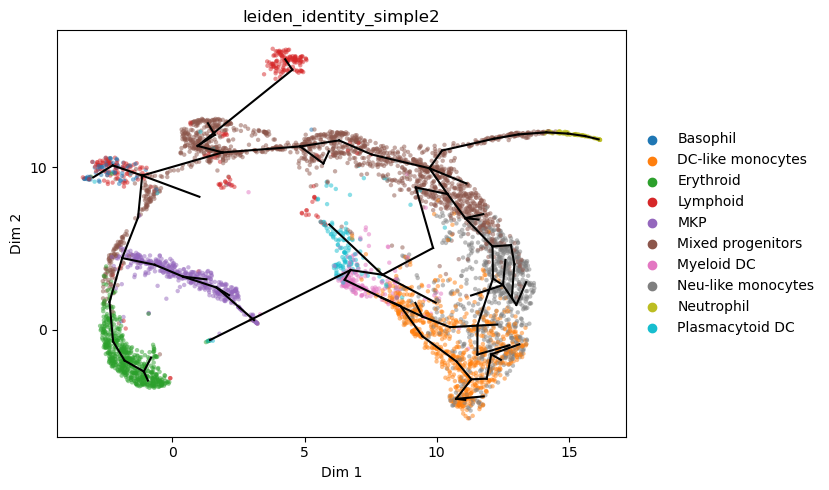
[29]:
st2.tl.learn_graph(a_s,n_nodes=100,epg_lambda=0.02,epg_mu=0.05,epg_alpha=0.02,verbose=1,store_evolution=True)
#max_candidates = {"AddNode2Node": 10,"BisectEdge": 10,"ShrinkEdge": 0,})
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5))
Constructing tree 1 of 1 / Subset 1 of 1
Computing EPG with 100 nodes on 5000 points and 14 dimensions
BARCODE ENERGY NNODES NEDGES NRIBS NSTARS NRAYS NRAYS2 MSE MSEP FVE FVEP UE UR URN URN2 URSD
4|17||81 8.6429 81 80 33 17 0 0 6.072 5.9942 0.8758 0.8774 2.5113 0.0597 4.8363 391.7442 0
3|18||82 8.5494 82 81 35 18 0 0 6.0528 5.9804 0.8762 0.8777 2.4401 0.0565 4.637 380.2305 0
2|19||83 8.451 83 82 37 19 0 0 6.0332 5.9652 0.8766 0.878 2.3627 0.0551 4.5701 379.3221 0
1|20||84 8.3884 84 83 39 20 0 0 6.0106 5.9425 0.8771 0.8785 2.321 0.0568 4.7691 400.6011 0
1|19||85 8.3177 85 84 42 19 0 0 5.9905 5.9232 0.8775 0.8788 2.2717 0.0555 4.7141 400.6955 0
20||86 8.2488 86 85 44 20 0 0 5.9737 5.908 0.8778 0.8792 2.2199 0.0552 4.7432 407.9185 0
19||87 8.1799 87 86 47 19 0 0 5.9594 5.8963 0.8781 0.8794 2.1685 0.052 4.5215 393.3734 0
18||88 8.1456 88 87 50 18 0 0 5.9684 5.905 0.8779 0.8792 2.1217 0.0555 4.8835 429.7443 0
17||89 8.0872 89 88 53 17 0 0 5.9558 5.8941 0.8782 0.8794 2.0789 0.0524 4.6658 415.2524 0
16||90 8.0388 90 89 56 16 0 0 5.9549 5.8942 0.8782 0.8794 2.0317 0.0522 4.6985 422.8661 0
15||91 8.0027 91 90 59 15 0 0 5.9558 5.8958 0.8782 0.8794 1.9936 0.0533 4.8512 441.456 0
14||92 7.9597 92 91 62 14 0 0 5.9475 5.8889 0.8784 0.8796 1.9568 0.0553 5.0916 468.43 0
13||93 7.9538 93 92 65 13 0 0 5.9667 5.9098 0.878 0.8791 1.9219 0.0651 6.0563 563.238 0
13||94 7.918 94 93 66 13 0 0 5.9639 5.9085 0.878 0.8791 1.8932 0.0608 5.7199 537.666 0
12||95 7.8795 95 94 69 12 0 0 5.9659 5.9112 0.878 0.8791 1.8541 0.0595 5.6491 536.6651 0
12||96 7.8339 96 95 70 12 0 0 5.9546 5.9009 0.8782 0.8793 1.8237 0.0556 5.3356 512.2143 0
11||97 7.8004 97 96 73 11 0 0 5.9618 5.9089 0.8781 0.8791 1.7868 0.0519 5.0353 488.4195 0
11||98 7.7592 98 97 74 11 0 0 5.937 5.8848 0.8786 0.8796 1.7718 0.0504 4.9379 483.9173 0
11||99 7.73 99 98 75 11 0 0 5.9231 5.8732 0.8789 0.8799 1.7558 0.0511 5.0544 500.3827 0
11||100 7.7023 100 99 76 11 0 0 5.9142 5.8656 0.879 0.88 1.7378 0.0503 5.0299 502.9927 0
127.9997 seconds elapsed
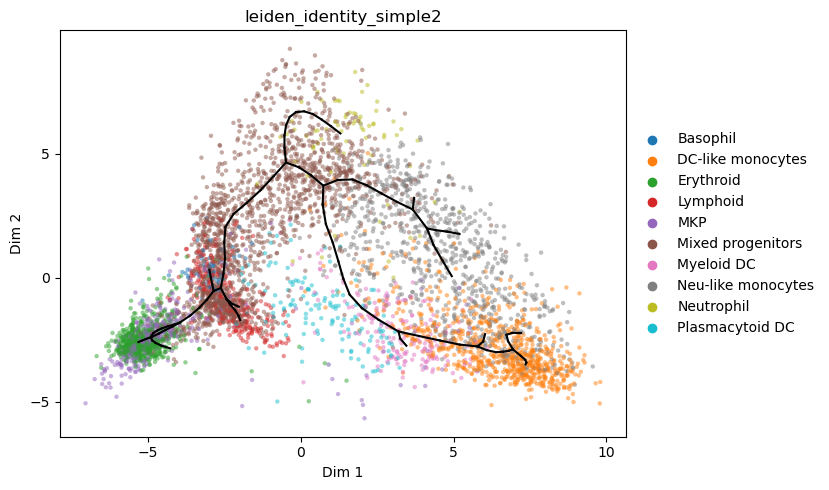
[30]:
st2.tl.project_graph(a_s, to_basis='X_umap', key='epg')
st2.pl.graph(a_s,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5),key='epg_from_X_dr_to_X_umap')
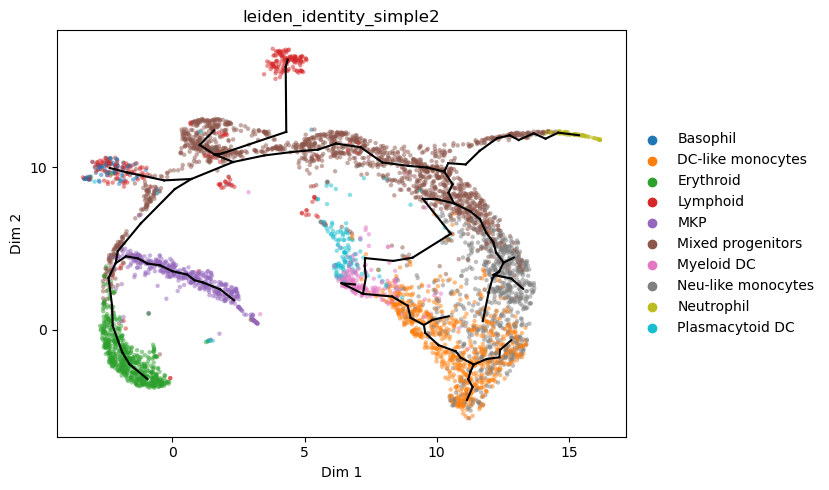
We can fine-tune the graph structure to: - add putative path from mixed progenitors to plasmacytoid DC - better fit paths for monocyte subtypes which are hard to capture - extend leaves of the graph to the very endpoints of the data distribution
[31]:
a_s2=a_s.copy()
st2.tl.del_path(a_s2,48,8)
st2.tl.add_path(a_s2,45,29)
st2.tl.add_path(a_s2,39,47,n_nodes=4)
st2.tl.add_path(a_s2,52,68)
st2.tl.del_path(a_s2,20,36)
st2.tl.del_path(a_s2,39,6)
st2.tl.del_path(a_s2,98,48)
st2.tl.del_path(a_s2,26,10)
st2.tl.del_path(a_s2,82,15)
st2.tl.add_path(a_s2,82,55)
st2.tl.refit_graph(a_s2,shift_nodes_pos={24 : a_s2.uns['epg']['node_pos'][24],# enforce same endpoint for monocytes
26 : a_s2.uns['epg']['node_pos'][24],})
st2.tl.extend_leaves(a_s2,LeafIDs=np.array([18,12,98,45,48,24,50,32]),Mode="QuantDists",ControlPar=.7)
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
Performing simulated annealing. This may take a while
(8, 14)
(103, 14)
[32]:
st2.tl.project_graph(a_s2, to_basis='X_umap', key='epg')
st2.pl.graph(a_s2,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(16,10),show_text=1)
st2.pl.graph(a_s2,color=['leiden_identity_simple2'],alpha=.5,size=10,fig_size=(8,5),key='epg_from_X_dr_to_X_umap')
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/elpigraph/utils.py:403: RuntimeWarning: invalid value encountered in divide
proj = (np.dot(X_embed.T, R) / R.sum(axis=0)).T
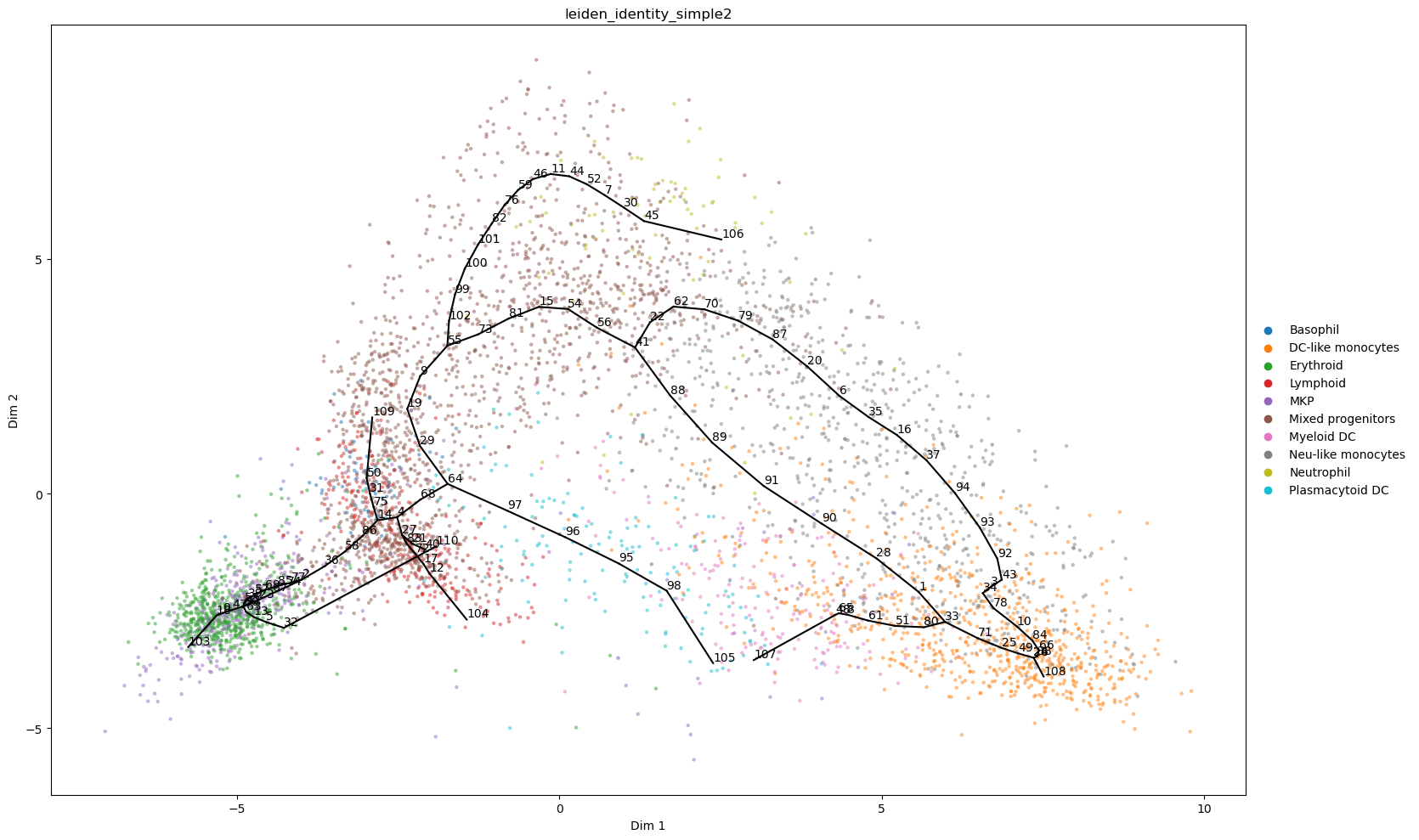
Now that we have the final graph we can compute pseudotime for further analyses
[33]:
adata_integrated.uns=a_s2.uns.copy()
st2.tl._elpigraph._store_graph_attributes(adata_integrated,adata_integrated.obsm['X_pca_transp_emd'],'epg')
st2.tl.infer_pseudotime(adata_integrated,40)
We can obtain a nicer 2D representation of nodes, e.g. by recomputing a UMAP directly including them
[34]:
# dummy anndata
a=sc.AnnData(np.concatenate((adata_integrated.obsm['X_pca_transp_emd'],adata_integrated.uns['epg']['node_pos'])))
sc.pp.neighbors(a,n_neighbors=50,metric='cosine')
sc.tl.umap(a,min_dist=0.05)
# store results
adata_integrated.obsm['X_umap_w_nodes']=a.obsm['X_umap'][:len(adata_integrated)]
adata_integrated.uns['umap_nodes']=a.obsm['X_umap'][len(adata_integrated):]
# plot
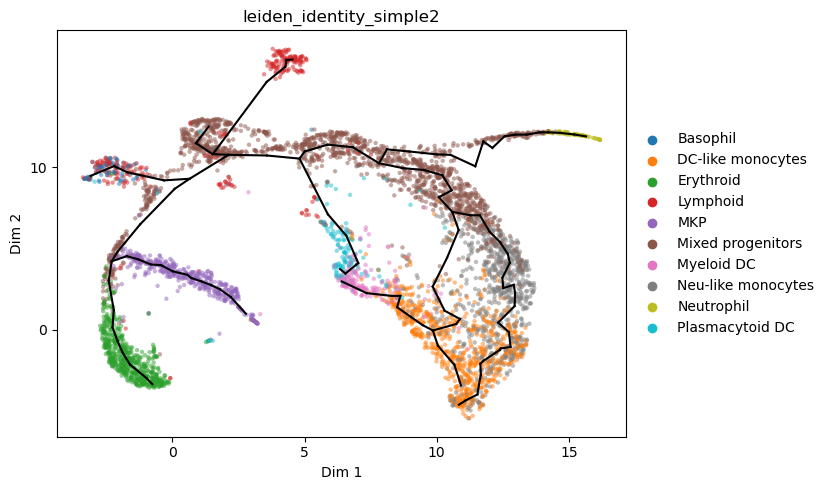
ax=plt.subplot()
_=sc.pl.embedding(adata_integrated,'X_umap_w_nodes',size=3,alpha=.5,
color='leiden_identity_simple2',show=False,return_fig=False,ax=ax)
ax.set_title(None)
ax.plot(adata_integrated.uns['umap_nodes'][adata_integrated.uns['epg']['edge'].T,0],
adata_integrated.uns['umap_nodes'][adata_integrated.uns['epg']['edge'].T,1], c='k',linewidth=2)
plt.show()
/tmp/ipykernel_2000/3214916952.py:2: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
a=sc.AnnData(np.concatenate((adata_integrated.obsm['X_pca_transp_emd'],adata_integrated.uns['epg']['node_pos'])))
/home/jo/anaconda3/envs/stream2/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(

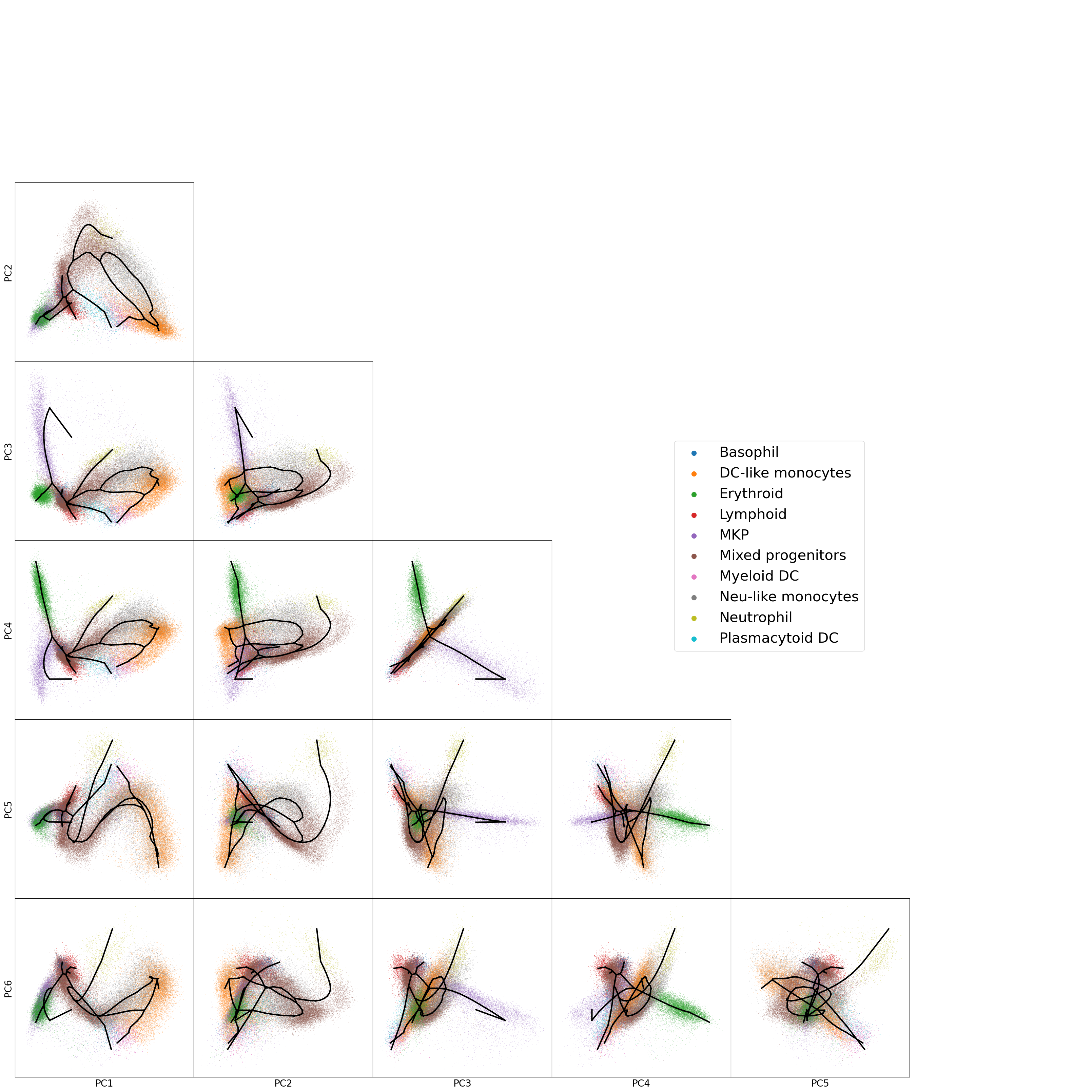
We can also plot various combination of PCs to get a better high-dimensional picture
[35]:
n_comps=6
f,axs=plt.subplots(n_comps,n_comps,figsize=(32,32),sharex='col',sharey='row')
for i in range(n_comps):
for j in range(i+1,n_comps):
_=sc.pl.embedding(adata_integrated,'X_pca_transp_emd',dimensions=(i,j),size=3,alpha=.5,
color='leiden_identity_simple2',show=False,return_fig=False,ax=axs[j,i])
handles, labels = axs[j,i].get_legend_handles_labels()
axs[j,i].get_legend().remove()
axs[j,i].set_title(None)
axs[j,i].plot(adata_integrated.uns['epg']['node_pos'][adata_integrated.uns['epg']['edge'].T,i],
adata_integrated.uns['epg']['node_pos'][adata_integrated.uns['epg']['edge'].T,j],
c='k',linewidth=3)
if i==0:
axs[j,i].set_ylabel(f'PC{j+1}',fontsize=20)
else:
axs[j,i].set_ylabel(None)
if j==(n_comps-1):
axs[j,i].set_xlabel(f'PC{i+1}',fontsize=20)
else:
axs[j,i].set_xlabel(None)
if i<j:
axs[i,i].axis('off')
axs[i,j].axis('off')
axs[-1,-1].axis('off')
lgnd=f.legend(handles, labels, loc='center right',bbox_to_anchor=[.8, 0.5],prop={'size': 30})
for handle in lgnd.legendHandles:
handle.set_sizes([100.0])
plt.tight_layout(pad=0,w_pad=0)
plt.show()
/tmp/ipykernel_2000/2761055043.py:30: MatplotlibDeprecationWarning: The legendHandles attribute was deprecated in Matplotlib 3.7 and will be removed two minor releases later. Use legend_handles instead.
for handle in lgnd.legendHandles:
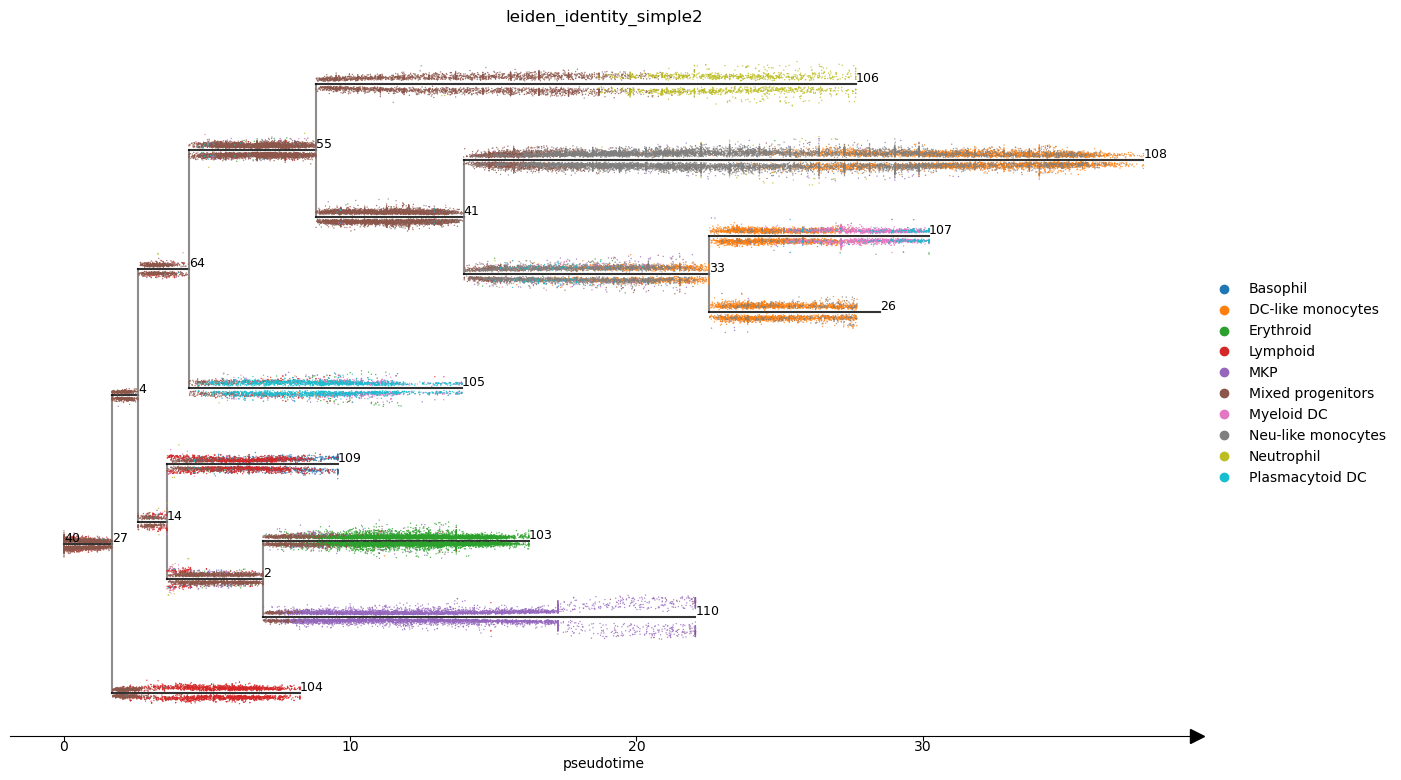
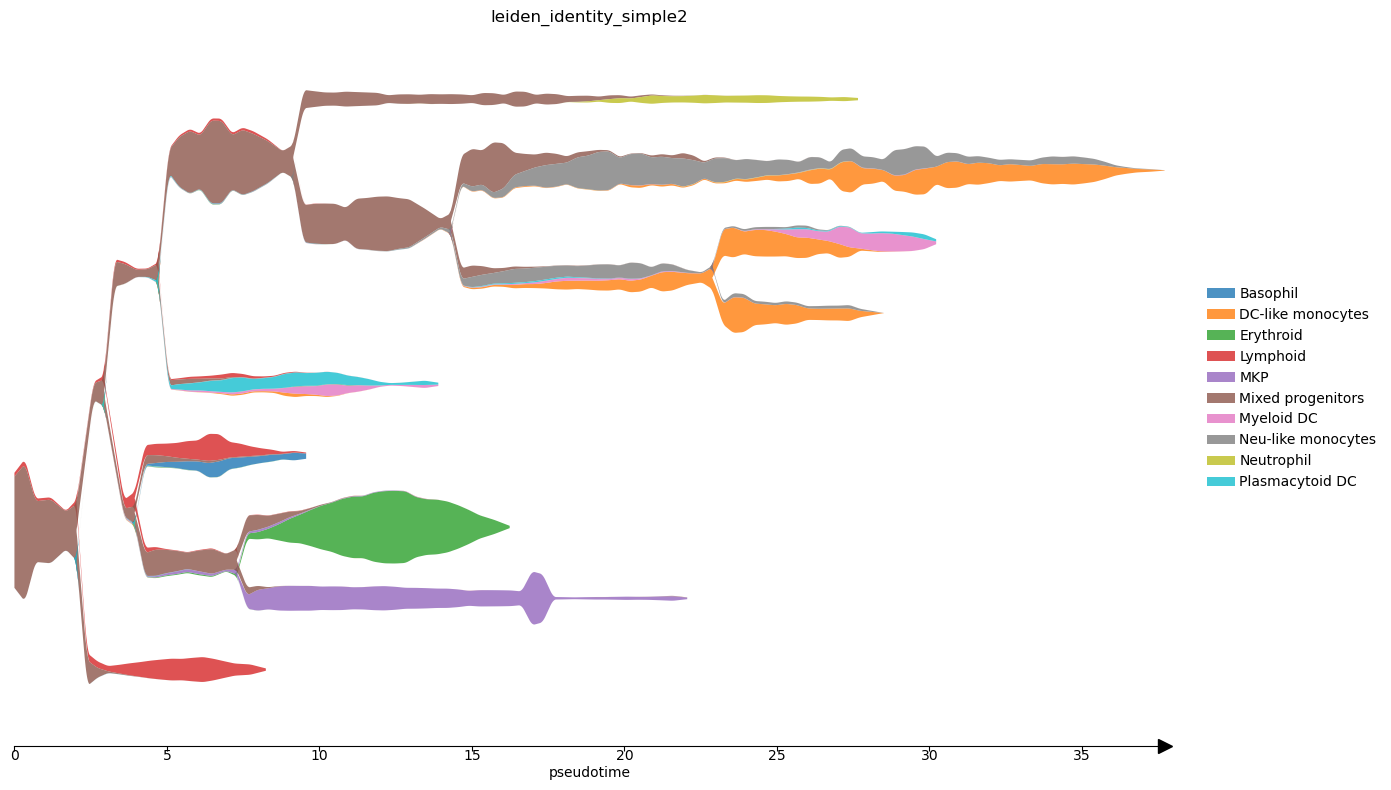
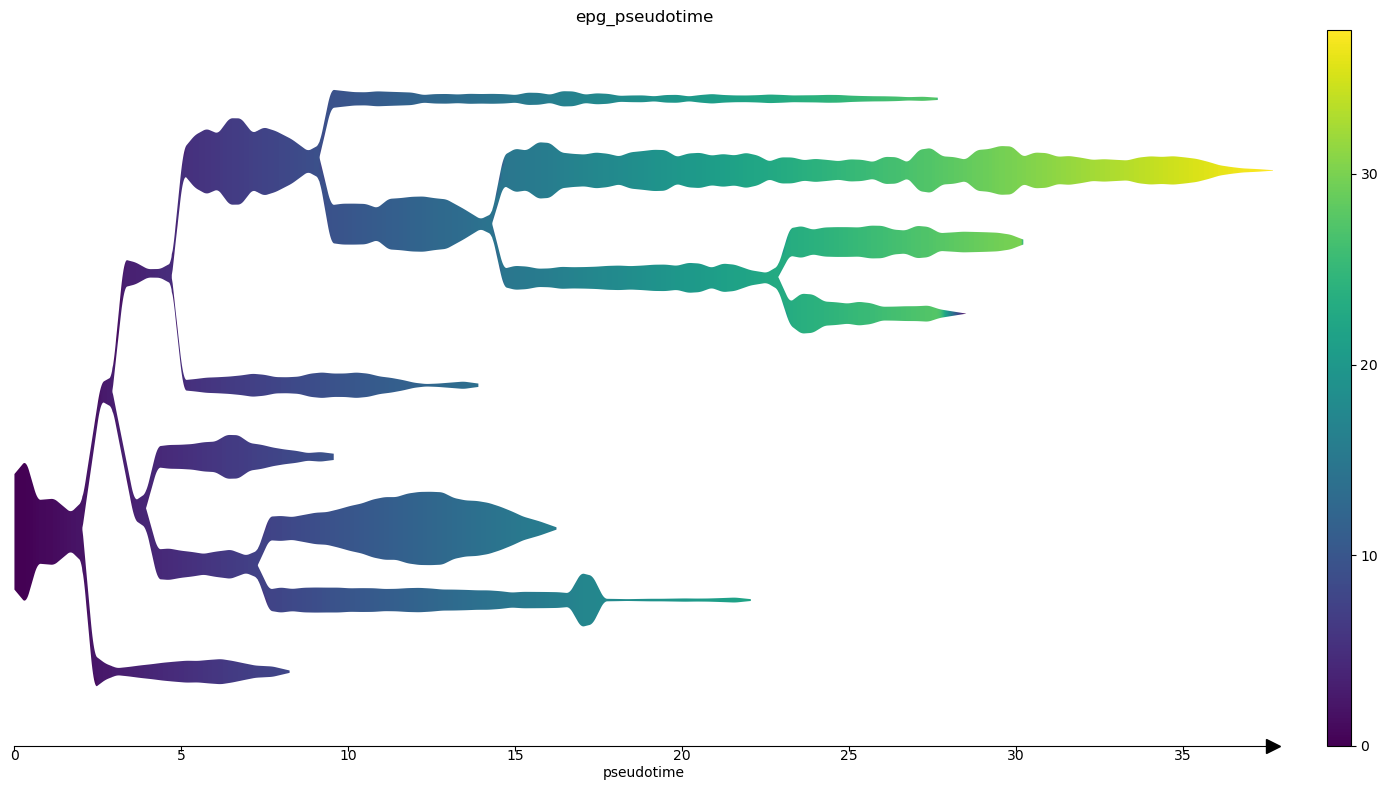
Finally we can produce STREAM plots.
[36]:
st2.pl.stream_sc(adata_integrated,40,dist_scale=.25,preference=[64],color=['leiden_identity_simple2'],fig_size=(14,8),size=1)
st2.pl.stream(adata_integrated,40,preference=[64],color=['leiden_identity_simple2'],fig_size=(14,8),dist_scale=2,factor_min_win=1.3,factor_num_win=4)
st2.pl.stream(adata_integrated,40,preference=[64],color=['epg_pseudotime'],fig_size=(14,8),dist_scale=2,factor_min_win=1.3,factor_num_win=4)
[ ]: